di Valeria Di Donna
Il Regolamento (UE) 745/2017 “MDR” sui dispositivi medici pone una grande attenzione al tema della sorveglianza post-commercializzazione.
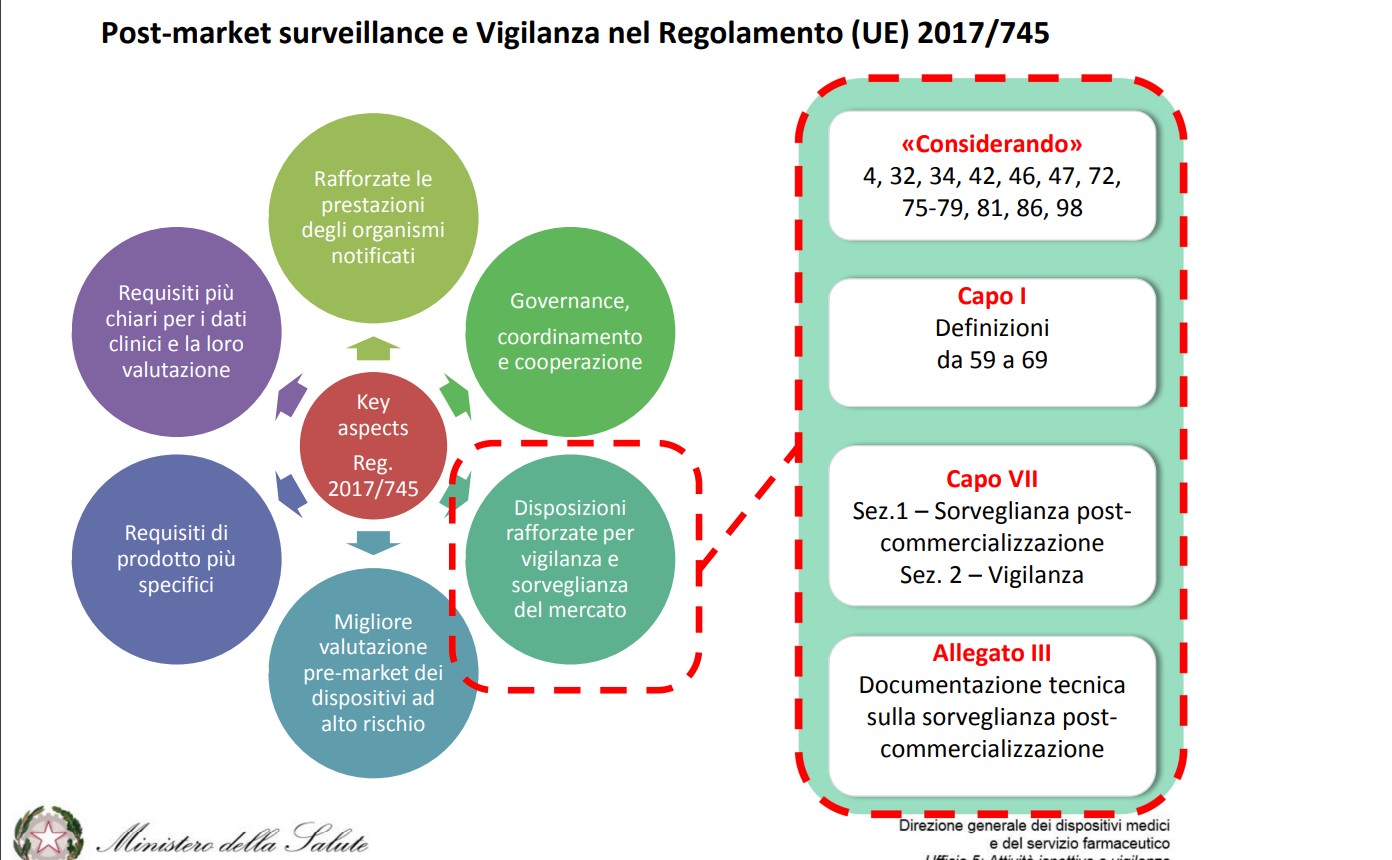
L’articolo 83, in particolare, definisce le caratteristiche del sistema di sorveglianza post-marketing (PMS), che deve essere specifico per il singolo dispositivo (sulla base delle sue caratteristiche) e proporzionato alla classe di rischio individuata dal fabbricante.
Il sistema PMS, inoltre, deve essere parte integrante del sistema di qualità del fabbricante. I dati raccolti in modo sistematico posso essere utilizzati per valutazioni sulla sicurezza, qualità ed efficacia dei dispositivi, lungo l’intero arco di vita e anche dopo la fine della commercializzazione.
Essi supportano anche l’aggiornamento della documentazione tecnica e del Riassunto della sicurezza e delle prestazione cliniche (SSCP), un nuovo tipo di documento previsto dal Regolamento con riferimento a dispositivi delle classi più alte, che deve venire validato da un organismo notificato e reso disponibile per mezzo della banca dati Eudamed dei dispositivi medici.
Le aziende sono chiamate a creare e aggiornare regolarmente anche il Piano di sorveglianza post -commercializzazione (piano PMS, art.84) e il Piano di Follow up Clinico post-commercializzazione (piano PMCF, art.61), quest’ultimo mirato ad approfondire in modo specifico i dati relativi alle destinazioni d’ uso di un certo dispositivo medico, con attenzione anche ai possibili utilizzi scorretti.
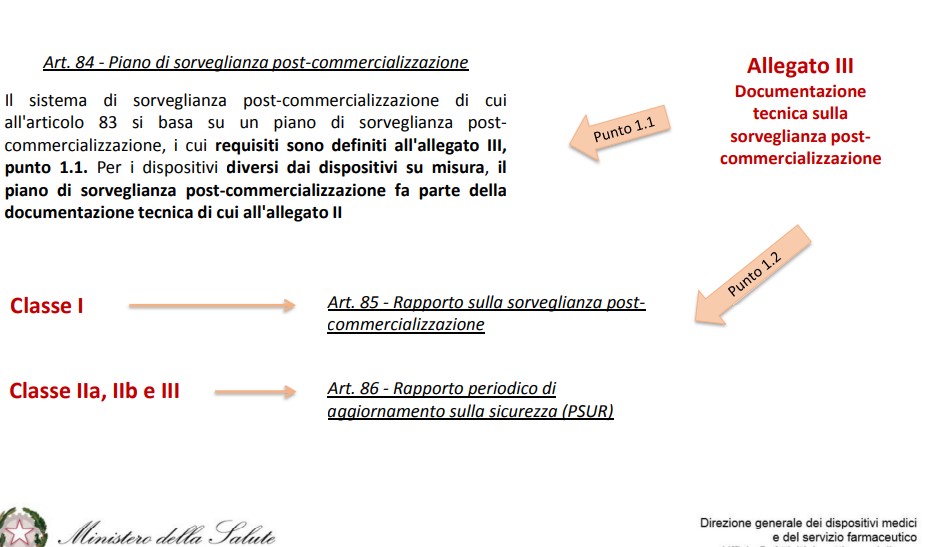
Il sistema di sorveglianza consta, quindi, di un piano PMS i cui requisiti sono definiti nell’allegato III e di un report, chiamato rapporto sulla sorveglianza post-commercializzazione (rapporto PMS) per DM di classe I oppure rapporto periodico di aggiornamento sulla sicurezza (o PSUR) per DM di classe IIa, IIb e III.
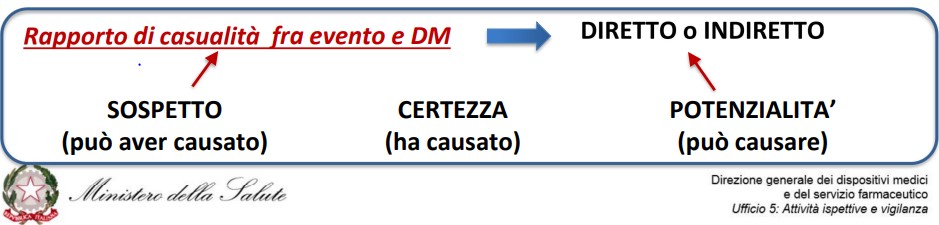
Il Regolamento ha anche rivisto la definizione di incidente grave: vi rientrano ora gli incidenti che, direttamente o indirettamente, abbiano causato, possano aver causato o possano causare il decesso o il grave deterioramento (anche solo temporaneo) delle condizioni di salute del paziente, dell’utilizzatore o di una altra persona, ovvero una grave minaccia per la salute pubblica.
Fonte: Notiziario Chimico Farmaceutico (NCF), marzo 2022
Per info:
Tel. 081.1890.2885
[/fusion_text][/fusion_builder_column][/fusion_builder_row][/fusion_builder_container]